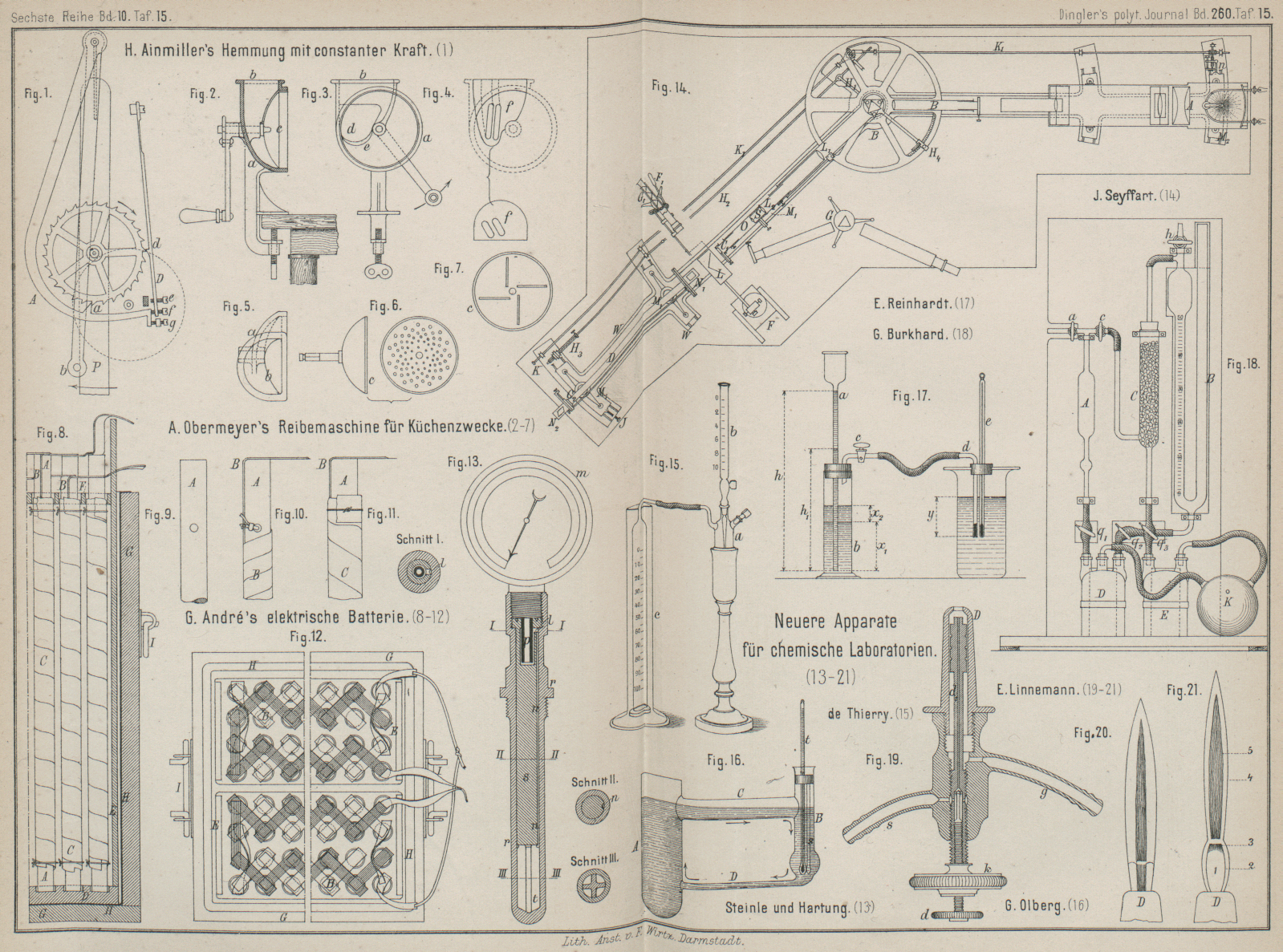
| Titel: | Neuere Apparate für chemische Laboratorien. |
| Fundstelle: | Band 260, Jahrgang 1886, S. 215 |
| Download: | XML |
Neuere Apparate für chemische
Laboratorien.
(Patentklasse 42. Fortsetzung des Berichtes Bd.
258 S. 500.)
Mit Abbildungen im Texte und auf Tafel 15.
Neuere Apparate für chemische Laboratorien.
Zur Bestimmung des wirksamen Sauerstoffes in
Wasserstoffsuperoxydlösungen läſst M. de
Thierry (Comptes rendus, 1886 Bd. 192 * S.
611) 10cc der zu prüfenden Flüssigkeit aus der
Röhre b (Fig. 15 Taf. 15) in das
mit Braunstein beschickte Entwickelungsgefäſs o
abflieſsen. Der sich entwickelnde Sauerstoff wird in der Glocke c über Wasser aufgefangen und das abgelesene Volumen in
bekannter Weise auf Normalbarometerstand und Temperatur umgerechnet.
Bei dem zerlegbaren Thermometer von Steinle und
Hartung in Quedlinburg (* D. R. P. Nr. 34328 vom 21. Juli
1885) ist, wie aus Fig. 13 Taf. 15 zu
entnehmen, das unten geschlossene Rohr r cylindrisch
ausgebohrt und an seinem oberen Ende mit Gewinde versehen, welches zum Einschrauben
des Einsatzstiftes s und des Manometers m dient. Der Stift s ist
an seinem oberen Ende mit einer Höhlung, in welche das Röhrchen p eintaucht, versehen; dieses steht durch das Loch l und die seitlich eingeritzte Rinne n in Verbindung mit dem unteren Theile t des Rohres r. An dieser
Stelle ist der Stift s mit den im Schnitte III
gezeichneten Aussparungen versehen. Soll das Thermometer in Benutzung genommen
werden, so füllt man das Rohr r mit einem flüssigen
Stoffe, schiebt den Stift s hinein und schraubt
denselben und hierauf das Manometer fest. Letzteres kann mit einer Flüssigkeit
gefüllt werden, welche sich weniger ausdehnt als die im Rohre befindliche
Flüssigkeit. Der durch das Röhrchen p gebildete
Wassersack verhindert ein etwaiges Vermischen der verschiedenen Flüssigkeiten.
Die Form des unteren Theiles des Stiftes (Schnitt III) ist deshalb gewählt, damit die
dort eingeschlossene Flüssigkeit schneller die Temperatur des Rohres r annimmt. Beim Füllen des Thermometers müssen alle
Bestandtheile desselben eine bestimmte Temperatur haben, wenn die Angaben des
Manometers, welches mit einer entsprechenden Temperaturskala versehen ist, richtig
sein sollen.
Zur Bestimmung des Schmelzpunktes von Fetten verwendet
C. Reinhardt (Zeitschrift
für analytische Chemie, 1885 * S. 17) ein mit einer Gradtheilung versehenes
Trichterrohr a (Fig. 17 Taf. 15) in einem
mit Gummistopfen verschlossenen Stehcylinder b. Die zur
Capillare ausgezogene Glasröhre d enthält eine etwa
20mm hohe Fettschicht. Man bringt vorerst
durch Einblasen von Luft durch den Hahn c das Wasser im
Trichterrohre a zum Steigen. Den Hahn c, welcher nunmehr verschlossen wird, verbindet man mit
dem Capillarrohre d mittels eines Gummischlauches.
Sodann öffnet man den Hahn c, liest die Höhen h und x1 ab, während der Höhenunterschied y gemessen wird.
Es wird jetzt wie gewöhnlich das Becherglas langsam erwärmt bis zur eintretenden Schmelzung des
Fettes, welch letzteres als Cylinderchen aus dem Capillarrohre herausgepreſst wird.
In diesem Augenblicke wird die Temperatur abgelesen. Das im Druckrohre a befindliche Wasser fällt bis nach h1, steigt indessen im
Stehcylinder um die Höhe x2. Der Wasserdruck p, unter welchem das Fett
herausgepreſst wurde, ist somit: p = h – (x1 + x2 + y) oder p = h – h1.
Je höher der Druck p gewählt wird, desto niedriger fällt
die Schmelzpunktbestimmung aus. Auch hat die lichte Weite des das Fett enthaltenden
Röhrchens d Einfluſs. Es betrug z.B. bei Paraffin bei
Anwendung eines 4mm weiten Röhrchens und bei einem
Wasserdrucke von 11 bis 13cm der Schmelzpunkt 52°,
bei Anwendung eines Capillarröhrchens und bei demselben Wasserdrucke aber 54° und
55°.
Man verwende zweckmäſsig stets ein und dasselbe Becherglas und fülle dasselbe immer
mit der gleichen Menge Wasser, was durch Anbringen einer Marke am Glase leicht
erreicht werden kann. Das Quecksilbergefäſs des in Zehntelgrad getheilten
Thermometers e und das im Röhrchen befindliche Fett
soll immer möglichst gleich weit vom Boden des Becherglases entfernt sein. Die
Fettschicht soll die Höhe des Quecksilbergefäſses nicht übersteigen; für längere
Fettschichten empfiehlt sich daher eine Ausdehnung der Schicht in wagerechter
Richtung, durch kreisförmige Biegung des das Fett enthaltenden Röhrchens. Die
Glaswandung des Fettröhrchens und diejenige des Thermometergefäſses soll zweckmäſsig
gleiche Stärke besitzen. Das allmähliche Erwärmen des Becherglases geschieht am
besten auf einer Asbestplatte. Capillar- oder 4mm-Röhrchen müssen nach der Füllung 1 bis 2 Tage lang liegen bleiben, ehe die
Schmelzpunktbestimmung vorgenommen werden kann, sonst erhält man zu niedrige
Angaben.
Das zu prüfende Fett filtrirt man durch ein trocknes Filter, am besten im Luftbade,
in ein kleines Becherglas, ohne jedoch den verbleibenden Rückstand mit auf das
Filter zu bringen. In einem kleinen Becherglase wird das filtrirte Fett
erforderlichen Falles geschmolzen, so daſs eine Fetthöhe von 10 bis 15mm erhalten wird. In das geschmolzene Fett taucht
man das 4mm weite Röhrchen ein, hält sodann
letzteres oben mittels des Zeigefingers zu, hebt das Röhrchen heraus, taucht die
untere Fläche desselben in kaltes Wasser, wartet einen Augenblick, läſst nun den
Finger los und stellt zum völligen Erstarren hin.
Für Schmelzpunktbestimmungen empfiehlt G. Olberg im Repertorium für
analytische Chemie, 1886 * S. 94 ein Oelbad,
in welchem ein gleichmäſsiger Kreislauf des Oeles ohne den ermüdenden und die
Genauigkeit der Bestimmung beeinflussenden Gebrauch des Rührers stattfindet.
Das 15cm lange Rohr A
(Fig. 16
Taf. 15) ist unten mit dem Rohre B mittels einer engen
Röhre D verbunden, oben durch die weitere Röhre C. Man füllt den Apparat so weit mit Oel, daſs dieses
beim Erhitzen den oberen Rand der Verbindungsröhre C
nicht ganz erreichen kann. Erhitzt man nun das Oel in A, so wird
ein Kreislauf des Oeles in der Pfeilrichtung stattfinden.
Zur Schmelzpunktbestimmung fügt man in das nach oben verlängerte Ende von B einen doppelt durchbohrten Kork ein, welcher
Thermometer t und Schmelzröhrchen s trägt. Die Verlängerung von B um einige Centimeter ist nöthig, da der Kork einige Grad des
Thermometers verdeckt, welche man dadurch sichtbar machen kann, daſs man den Kork
verschiebt, während das Thermometer bis zur bestimmten Tiefe eingetaucht bleibt. Es
ist von Vortheil, Thermometer und Schmelzröhre möglichst tief in den Apparat
einzuführen, da sich der Temperaturwechsel immer ruhiger vollzieht, je mehr man sich
von der Oberfläche entfernt. Auſserdem stellt man Thermometer und Schmelzröhrchen
praktisch so, daſs die Ebene beider zur Richtung der Röhre C senkrecht steht, weshalb man den Apparat so aufstellt, daſs von A und B der Cylinder B dem Beobachter am nächsten steht. Um Thermometer und
Schmelzröhrchen dem Einflüsse der kühleren Glaswände möglichst zu entziehen, bläst
man an der Stelle, wo die Kugel des Thermometers sich befindet, B etwas weiter.
Zur Bestimmung der Kohlensäure in Saturationsgasen will
G. Burkhard (Neue
Zeitschrift für Rübenzuckerindustrie, 1886 Bd. 16 * S. 115) die Messung des
Gases vor und nach der Behandlung mit Kalilauge in zwei gesonderten Meſsröhren
vornehmen. Die Entfernung der Kohlensäure durch Kalilauge erfolgt in einem zwischen
beide Rohre eingeschalteten Gefäſse, in welchem das aus dem ersten Rohre verdrängte
Gas von Kohlensäure befreit in das zweite Meſsrohr gelangt.
Der in Fig. 18
Taf. 15 dargestellte Apparat besteht aus zwei getheilten Röhren A und B und dem zwischen
beide eingeschalteten Absorptionsrohre C. Die
Vollpipette A trägt ober- und unterhalb des Körpers
eine Marke und dient zum Abmessen des zu untersuchenden Gasvolumens. Der linke
Schenkel des Rohres B faſst zwischen den Marken 0 und
100 genau so viel Gas wie A zwischen seinen beiden
Marken. Unterhalb des Glashahnes h ist seitlich eine
Röhre angeschmolzen, welche B mit dem Absorptionsrohre
C verbindet. An die Pipette A ist über der oberen Marke ein enges T-Stück angeschmolzen, dessen
Schenkel die Glashähne a und c tragen; ersterer vermittelt die Zuführung bezieh. den Abschluſs des zu
untersuchenden Gases; Hahn c stellt Abschluſs und
Verbindung von A mit dem Absorptionsgefäſse C her.
Beide Meſsröhren münden nach unten mittels Gummischläuchen, welche mit Quetschhähnen
q1 und q2 versehen sind, bis
auf den Boden einer dreifach tubulirten Flasche D. Die
Flasche enthält Wasser, welches mit Kohlensäure gesättigt ist und als
Sperrflüssigkeit für die Meſsröhren A und B dient. Das Absorptionsrohr C ist mit kleinen Bimssteinstücken oder Glaskugeln angefüllt. Am Boden
steht C durch ein engeres Glasrohr mit Gummischlauch
und Quetschhahn q3 mit
einer zweifach tubulirten Flasche E luftdicht in Verbindung, welche Kalilauge
enthält; der andere Tubus der Flasche E und der dritte
der Flasche D sind mittels Gummistopfen und
Winkelröhren mit den beiden Schenkeln des bekannten, aus Kautschuk hergestellten
Druckballens K verbunden. Durch Oeffnen der
Quetschhähne und Drücken auf K, während man dessen
kleine Oeffnung mit dem Daumen verschliefst, kann man leicht in die Rohre A und B Wasser aus D und ins Rohr C Kalilauge
aus E bis zu jeder beliebigen Höhe hinübertreiben. Vor
jeder Gasuntersuchung wird nun durch diese Vorrichtung Kalilauge in C bis nahe zum oberen Rande hinaufgedrückt, so daſs
sämmtlicher Bimsstein mit derselben befeuchtet wird, und der Ueberschuſs der Lauge
wieder nach E abgelassen. Man hat hierdurch eine
groſse, mit Kalilauge bedeckte Oberfläche erzeugt, welche Kohlensäure sehr rasch
absorbirt.
Die Ausführung einer Bestimmung der in einem Gase enthaltenen
Kohlensäure geschieht nun in folgender Weise: Bei offenen Hähnen füllt man die
Meſsröhren B und A nach
einander durch Drücken auf K und Oeffnen der
Quetschhähne q2 bezieh.
q1 mit Wasser an
und zwar derart, daſs man zuerst B bis genau zur
100-Marke einstellt, in A aber dann das Wasser bis in
die Hähne a und c
hineintreibt und in dem Augenblicke, wo das Wasser c
trifft, q1 und hierauf
c schlieſst, während Hahn a offen bleibt. Zugleich ist hierdurch auch der Rohransatz bei a vollständig mit Wasser gefüllt worden. Indem die
Glashähne in dieser Stellung verbleiben, d.h. indem a
und h offen, c geschlossen
ist, wird nun durch Oeffnen von q3 und Drücken auf K
Kalilauge in C bis nahe zum oberen Rande
hineingetrieben und dann wieder abgelassen, wodurch der in C enthaltene Bimsstein mit Lauge vollständig durchtränkt zurückbleibt. Man
läſst nun das zu prüfende Gas wenige Augenblicke aus dem Zuleitungsschlauche frei
ausströmen, verbindet denselben hierauf mit dem Glasstutzen bei a, öffnet q1 und läſst das Gas, welches das Wasser aus A vor sich her treibt, bis unterhalb der kleinen Kugel
von A dringen. Man schlieſst hierauf q1 und dann a und stellt das Wasser mit Hilfe von K genau auf die untere Marke von A ein. Durch den in der Gaszuleitung vorhandenen Druck
und durch das Hinaufdrücken des Wassers bis zur unteren Marke von A ist ein gewisser Ueberschuſs von Gas in A hervorgerufen worden. Derselbe wird durch Oeffnen des
Hahnes c entfernt und dient zu gleicher Zeit dazu, das
enge Glasrohr zwischen c und C gleichfalls mit dem zu untersuchenden Gasgemische anzufüllen. Dieser
Zweck wird noch dadurch um so vollkommener erreicht, als das Gasgemisch die kurze
Wassersäule, welche sich noch vor C im T-Stück befand,
vor sich her treibt, somit sich also nicht mit der in dem Glasröhre hinter c befindlichen Luft vermischen kann. Indem Hahn c offen bleibt, schlieſst man nun auch h und drückt langsam mit Hilfe von K bei geöffnetem Quetschhahne q1 Wasser in A hinein; das eindringende Wasser treibt das entsprechende Volumen Gas vor
sich her nach C hinüber, während das Wasser im rechten
Schenkel von B steigt und sein Ueberschuſs in kurzen
Zwischenräumen durch q2
abgelassen wird. Man läſst das Wasser in A genau bis
zur oberen Marke steigen und schlieſst dann q1. Durch vorsichtiges Oeffnen von q2 stellt man nun die
beiden Schenkelrohre von B langsam zu gleicher Höhe ein
und schlieſst c. Hierdurch hat man genau 50cc Gas in C
hineingetrieben, wo dasselbe von Kohlensäure befreit und sein Volumen entsprechend
verringert wird. Nach einigen Minuten beobachtet man nochmals den Wasserstand in den
beiden Schenkeln von B, regelt nöthigen Falles durch
Hinaufdrücken von Wasser in das Druckrohr und liest nun an der procentischen
Theilung des linken Schenkels die Volumen-Procent Kohlensäure ab, welche das
Gasgemisch enthielt.
Zur Herstellung von Zirkonlicht verwendet E. Linnemann (Monatshefte für
Chemie, 1885 * S. 899) ein Leuchtgas-Sauerstoffgebläse. Nebenstehend ist die Lampe sammt
Zirkonblättchen z in ⅕ n. Gr. abgebildet. Fig. 19 Taf.
15 zeigt vergröſsert die eigentliche Brenndüse, welche im Inneren oben kegelförmig,
aber mit cylindrischer Bohrung ausläuft. Das Gras tritt durch Rohr g, Sauerstoff durch Rohr s
ein. Die Zuführungsröhre für Sauerstoff ist am oberen Ende cylindrisch, die
Ausfluſsöffnung des Gases genau anfüllend und starkwandig mit kaum nadeldicker
Ausfluſsöffnung. Das untere Ende der Röhre ist eine gasdichte Schraube, welche
mittels des rändrirten Kopfes k genau gegen die
Ausfluſsöffnung des Gases gestellt werden kann, wodurch zugleich eine Regelung des
Leuchtgasausflusses für verschiedene Druckverhältnisse und die eigentliche Formung
der Flamme bewirkt wird. Die Zuführungsröhre d1 für Sauerstoff trägt etwas unterhalb ihres oberen
Endes eine in den dort noch cylindrischen Theil der Brennerdüse D passende Erweiterung, welche mit 6 kleinen
Längsrinnen und 3 Querrinnen versehen ist. Diese Vorrichtung hat den Zweck, das
seitlich einströmende Gas vollständig gleichmäſsig zu vertheilen und im ganzen
Querschnitte der Ausfluſsöffnung unter gleichmäſsigem Drucke austreten zu
lassen.
Textabbildung Bd. 260, S. 219 Der Sauerstoff tritt aus dem Rohre s mit
Hilfe einer Aussparung in der Schraube d durch 4 feine
Oeffnungen in das Innere der Röhre d1. Da es zur richtigen Formung der Flamme nothwendig
ist, daſs der Sauerstoff an seiner Ausfluſsöffnung sogleich mit dem gehörigen
Ueberdrucke austritt, so ist es wünschenswerth, unmittelbar vor dem starren
Zuführungsrohre der Lampe ein kurzes Stück starkwandigen Kautschukschlauches mit
Niederschraubklemmen anzubringen, da durch kurzes Zusammendrücken desselben und
plötzliches Auslassen das gewünschte Ziel leicht und sicher erreicht wird.
Fig. 21 Taf.
15 zeigt die richtig geformte und ganz lautlos brennende kleinere Flamme mit der
etwa 1cm vor der Brennerdüse D liegenden, stark weiſsblau leuchtenden, heiſsesten
Stelle 3 der Flamme. Der Saum 1 ist wie der entsprechende Theil der Bunsenflamme dunkel, der Saum 2 kaum sichtbar blau, der Saum 4 etwas kräftiger blau und der innere Theil 5, die Verlängerung des brennenden Sauerstoffstromes, deutlich weiſslich blau
gefärbt. In dieser Flamme zeigt nur der sehr kleine heiſseste Theil 3 ein schön entwickeltes Kohlenstoffspectrum; die
übrigen Theile der Flamme senden kein merkbares Licht in den Spectralapparat.
Fig. 20 Taf.
15 zeigt die Flamme in der Form, welche den jetzt gebräuchlichen Knallgaslampen
entspricht, wobei der Sauerstoffstrom schon innerhalb der Düse zu brennen anfängt
und die Düse D sehr heiſs wird, wie bei einem
zurückgeschlagenen Bunsenbrenner. Die Wärmewirkung dieser Flamme, welche sich stets
bildet, wenn der Sauerstoff nicht den nöthigen Ueberdruck zeigt, ist ganz
unvergleichlich geringer. Ebenso leicht läſst sich mit dieser Lampe eine vollkommen
geräuschlos abbrennende Wasserstoff-Sauerstoffflamme (Knallgasflamme) erzeugen, bei
welcher der Sauerstoffstrom erst auſserhalb der Düse zu brennen anfängt und die Düse
nicht merkbar heiſs wird.
Beim Einführen einer Sodaperle in den heiſsesten Theil 3
(Fig. 21)
der Leuchtgas-Sauerstoffflamme entsteht ein so kräftiges Licht, daſs man den Glanz
desselben mit freiem Auge nicht zu ertragen vermag. Die so erzeugten Spectren der
Alkalimetalle sind von wundervoller Reinheit und vollständig entwickelt, d.h. es
treten in denselben wie bei der Hitze des elektrischen Flammenbogens alle Linien
auf, welche diese Metalle überhaupt entwickeln können. Die
Leuchtgas-Sauerstoffflamme selbst zeigt, wie schon erwähnt, nur im heiſsesten
Theile, auf der stark weiſsblau leuchtenden kurzen Stelle 3, ein selbstständiges Spectrum. Da sich nun in demselben Theile der
Flamme die weiſsglühende Perle der geschmolzenen Verbindung befindet, so muſs dieser
Theil der Flamme ohnedies abgeblendet werden, was am besten durch die mittels einer
Linse zu bewirkende zweckentsprechende Projection des Flammenbildes auf den Spalt
bewirkt werden kann. Durch diese Umstände erklärt sich die vollkommene Reinheit
dieser Spectren und der Ausschluſs von störenden Nebenspectren. Das Bild dieser
Spectren ist das Schönste unter allen erzeugbaren Spectren. Auf ganz dunklem Grunde
zeigt so das Lithium vier Linien, das Natrium, welches nur in der Umgebung der
glänzend leuchtenden Verbreiterungen der umgekehrten D-Linien hellen Hintergrund zeigt, ergibt 5 Doppellinien, das Kaliumspectrum
27 deutliche Linien.
Die Lampe eignet sich auch vortrefflich zur Erzeugung eines sehr starken Kalklichtes, zumal wenn man die Flamme so richtet, daſs
der weiſsblau leuchtende heiſseste Theil 3 gerade die
Kalkoberfläche berührt. Das entwickelte Licht ist auſserordentlich stark und für
kurze Zeit auffallend ruhig, nimmt aber bald ab, weil die auſserordentlich
concentrirte Hitze der Flamme den Kalk schmilzt. Da Magnesia noch leichter in der Hitze dieser Flamme schmilzt als Kalk, so
wurde Zirkonerde versucht. Die Schwierigkeit,
Zirkonerde in dichten Stücken zu erhalten, beruht darauf, daſs die Erde für sich
eine amorphe, absolut unschmelzbare, pulverförmige Masse darstellt und daſs deren
Verbindungen, an der Luft geglüht, ausnahmlos, ohne zu schmelzen oder zu sintern,
unter Zersetzung pulverförmige Erde als Rückstand lassen.
Die zahlreichen Versuche, welche zur Herstellung fester Massen von Zirkonerde
unternommen wurden, ergaben zunächst, daſs jeder als Fluſsmittel gedachte Zusatz zur
Zirkonerde die Schwierigkeit nur erhöht und daſs man nur zum Ziele gelangt, wenn man
ganz chemisch reine Zirkonerde, namentlich frei von Alkalien und alkalischen Erden,
verwendet. Zur Erzeugung von Zirkonlicht benutzt E.
Linnemann die Zirkonerde in Form von Scheiben, welche 15mm im Durchmesser und 4mm Dicke besitzen und in einen kleinen Teller von
nicht zu dünnem Platinblech gefaſst sind, der seinerseits einen Platindraht trägt,
um das Ganze zweckentsprechend an der Lampe befestigen zu können, wie die Textfigur
ersehen läſst.
Zunächst wird reines Zirkonchlorid in nicht zu groſser Menge in bedecktem
Porzellantiegel im Gasofen anhaltend erhitzt, wobei schneeweiſse Zirkonerde
zurückbleibt. Diese wird im Achatmörser zum feinsten Pulver zerrieben und in einer
zur Erzeugung von etwa 3 bis 4mm dicken Blättchen
erforderlichen Menge in einen Stahlmörser von etwa 15mm Durchmesser, wie solche zum Zerkleinern von Mineralien im Gebrauche
sind, eingeschüttet. Das durch Klopfen gleichmäſsig ausgebreitete Pulver wird mit
Hilfe des Stahlstempels erst sachte mit der Hand, dann möglichst fest
zusammengepreſst, worauf die Scheibchen durch ruhigen Schraubendruck aus der Stanze
herausgedrückt werden. Die so gewonnenen Scheibchen sind so weit haltbar, daſs sie
sich vorsichtig anfassen lassen, ohne zu brechen. Ihre weitere Haltbarkeit und
Härtung erhalten die Zirkonerdeblättchen durch bloſses anhaltendes, allmählich immer
heftigeres Erhitzen, zuletzt im Knallgasgebläse. Hierbei findet ein theilweises
Sintern unter Volumenverminderung statt, wobei die Blättchen in Folge
ungleichmäſsigen Schwindens häufig in mehrere Stücke zerspringen. Eine Vorrichtung,
welche eine gleichmäſsigere Erhitzung im Knallgasgebläse zulieſse, würde dieses
Springen wohl vermeiden lassen. Gesprungene Scheibchen werden aufs Neue im
Achatmörser aufs Feinste gepulvert, gepreſst und erhitzt. Die Scheibchen springen
jetzt schon viel seltener und meist nur in zwei Stücke. Bei neuerlicher Formung
bleibt nun das Scheibchen entweder ganz oder, wenn Sprünge entstehen, setzen sie
nicht mehr durch. Das Ausglühen des Zirkonerdeblättchens im Knallgasgebläse kann nur
auf Platinunterlage bewirkt werden, da dünne Lagen von Zirkonerde auf Thon z.B. wie
Wachs durchschmelzen.
Ein im Feuer ganz gebliebenes Zirkonerdescheibchen ist hinreichend hart, um in den
kleinen Platinteller gefaſst werden zu können. Von unverwüstlicher Dauer sind die so
gewonnenen Zirkonerdeblättchen allerdings auch nicht; sie blättern im Gebrauche
allmählich von der Oberfläche ab, zumal bei zu raschem Anheizen; allein man kann ein
und dasselbe Blättchen doch viele Hundert mal gebrauchen, bevor eine so groſse
Unebenheit der Oberfläche entsteht, daſs eine Neuformung der Scheibe nothwendig
würde.
Benutzt man die vollkommen lautlos, ganz ruhig und stetig brennende Flamme des
Leuchtgas-Sauerstoffgebläses, so hat man das Zirkonerdeblättchen so zu richten, daſs
der blaue Punkt 3 der Flamme gerade die Oberfläche der
Zirkonerde berührt. Obgleich fast das ganze Scheibchen weiſsglühend wird, ist es
doch nur eine kaum 5mm Durchmesser zeigende
kreisrunde Fläche, welche den höchsten Grad der Weiſsglut erreicht, woraus bei dem
erzielten bedeutenden Lichte eine auſserordentlich hohe Lichtstärke der
Flächeneinheit folgt. Entsprechend dieser groſsen Concentration des Lichtes erhält
man auch sehr scharf begrenzte Schatten. Das von dem glühenden Zirkonerdeblättchen
ausgehende sehr concentrirte, vollkommen ruhige und stetige Licht ist rein weiſs.
Bei spectraler Zerlegung gibt es ein continuirliches Spectrum, welches die Frauenhoffer'schen Linien A bis H umfaſst und keine Spur einer hellen
Spectrallinie aufweist, wie etwa das Kalklicht, welches
neben der Natriumlinie die rothen und grünen Kalkbänder zeigt. Dieser Umstand läſst
das Zirkonlicht als einen werthvollen Ersatz für Sonnenlicht erscheinen und es ist
deshalb für eine Reihe von Versuchen dem elektrischen Lichte vorzuziehen. Ein
weiterer Vortheil ergibt sich daraus, daſs die glühende Zirkonerde, wahrscheinlich
im Zusammenhange mit der erzeugten groſsen Lichtmenge, ganz auffallend wenig Wärme
ausstrahlt, so daſs die Lichtquelle den zu beleuchtenden Gegenständen sehr nahe
gebracht werden kann. Bei bezüglichen Versuchen betrug der Gasdruck im Mittel 6cm Wassersäule, der Druck des Sauerstoffes im
Mittel das 15 fache davon. Die beobachteten Lichtstärken reichten je nach dem
Verbrauche an Sauerstoff und Gas von 60 bis 280 Kerzen. Hierbei verlangten im Mittel
vieler Versuche etwa 60 Kerzen stündlich 24l
Leuchtgas und 15l Sauerstoffgas, 120 Kerzen 37l Leuchtgas und 26l Sauerstoffgas, 200 Kerzen 48l
Leuchtgas und 44l Sauerstoff. Hierbei ist zu
bemerken, daſs Lichtstärken von 60 bis 120 Kerzen noch mit der vollkommen
geräuschlos abbrennenden Flamme erzeugt werden, während höhere Lichtstärken nur mit
bereits pfeifender Flamme entstehen.
Das Dispersionspolarimeter von J.
Seyffart in Berlin (* D. R. P. Nr. 34339 vom 28. Juli
1885) ist mit Einstellung auf nur eine bestimmte Wellenlänge des Lichtes
zum Gebrauche in Zuckerfabriken, mit beliebiger Einstellung auf irgend welche
gewünschte Wellenlänge des Lichtes zum Gebrauche in wissenschaftlichen Laboratorien
bestimmt.
Der Lichterzeuger A (Fig. 14 Taf. 15) ist
entweder mit Vorrichtungen für Drummond's Kreidelicht
oder mit Erdölbrennern versehen. Der Spectralapparat B
dient zur Zerlegung des Lichtes in parallelschichtige monochromatische Farben. Der
eigentliche Polarisationsapparat D besteht aus zwei Nicol'schen Prismen N1 und N2, welche den Lichtstrahl in zwei polarisirte
Strahlen zerlegen, wovon der eine seitlich vernichtet wird, während nur der andere
das Prisma durchsetzt. Eines der beiden Prismen ist um meſsbare Winkel drehbar.
Zwischen diese beiden Prismen kommt die zu untersuchende Probe.
Das Eigenthümliche des Apparates besteht in der Einschaltung einer verstellbaren Wand
mit feinem Spalte S im beiderseitigen Brennpunkte des
Okulares O und der zweiten Objectivlinse L2 des Fernrohres vom
Spectralapparate, in systematischer Verbindung mit zwei Cylinder linsen, wovon die
erste C1 vor, der
Strahlrichtung nach hinter dem obengenannten Okular, mit ihrer Achse in gekreuzter
Stellung zu dem Spalte S sich befindet und achsial, im
Kreise sowie wagerecht verstellt werden kann, während die zweite Cylinderlinse C2 zwischen dem zweiten
Nicol'schen Prisma N2 und der Probelösung, wagerecht und im
Kreise verschiebbar, angebracht ist. Die Cylinderlinse C1 kann jedoch auch hinter dem Nicol N1 und die
Cylinderlinse C2 hinter
dem Nicol N2 befestigt
werden. Auch lassen sich die Cylinderlinsen durch schwach elliptische Linsen
ersetzen.
Wesentlich ist ferner ein verschiebbarer, einfacher oder doppelter Plan- oder
Winkelspiegel L, mit welchem man das aus dem
Spectralapparate austretende Lichtbündel nach einem seitlich angebrachten zweiten
Spectralapparate oder Spectroskope G und G1 hinablenken und aus
der Spectrallinie das monochromatische Lichtbündel auf seine mittlere Wellenlänge
genau prüfen kann. Während dieser Prüfung kann der Winkelspiegel hin und her bewegt
werden mittels des Ständerschlittens F oder einer mit
dem Spectroskope G1
verbundenen einfachen Handhabe F1, welche auch den Spiegel tragen kann. In Fig. 14 findet
sich auf der einen Seite des Polarimeters eine Anordnung mit verschiebbarem
Ständerschlitten F und Spectralapparat, auf der anderen
Seite eine solche mit einfachem Planspiegel, Spectroskop „à vision directe“ und Handhabe F1 angegeben.
Für den Gebrauch in Fabrikslaboratorien ist das Prisma des groſsen Spectralapparates
B parallel zu seiner Brechungskante etwas dreh- und
genau einstellbar. In der Zeichnung ist der drehbare Teller, welcher das dreikantige
Prismatischchen mit dem Prisma trägt, in fester Verbindung gedacht mit der die
Trägersäule des Spectralapparates durchdringenden Achse, welche an ihrem unteren
Ende einen Hebel H1
trägt, der durch die Triebstangen H2 und Schraube H3 bewegt und ausgeschaltet werden kann. Für den
wissenschaftlichen Gebrauch läſst sich das Prisma um genau meſsbare, an dem Nonius
H4 abzulesende
Winkel einstellen.
Für beide Gebrauchsweisen kann der groſse Spectralapparat statt mit einem einfachen
Prisma mit einem Systeme zusammengehöriger Prismen „à
vision directe“ versehen sein.
Der Polarisationsapparat 2), obwohl unabhängig vom Spectralapparate aufgestellt, kann
dadurch, daſs er auf einer bis unter den letzteren untergreifenden Schiene M1 steht, die
ihrerseits auf einer das gesammte Dispersionspolarimeter tragenden Grundplatte W entweder unmittelbar gleitet oder auf
Zwischenröllchen sich fortbewegt, um ein Weniges um die ideelle Drehungsachse vom
Prisma des Spectralapparates bewegt und genau eingestellt werden, wie bei J erkennbar ist.
Es kann auch die Lichtquelle des Skioptikons A um eben
dieselbe Achse bewegt und regulirt werden, entweder unmittelbar am Skioptikon
selbst, oder durch Verlängerungsspindeln nach dem Beobachter hin zur bequemen
Einstellung während der Beobachtung. In Fig. 14 ist eine
bezügliche Einrichtung mit verkuppelten Drehspindeln K1, Bewegungsschraube, Zahnrädchen,
Schraubenspindel und Schraubenmutter, welche durch ein bewegliches Glied n die Unterlagsschiene M2 für das Skioptikon hin und her schiebt,
sobald die Drehspindeln durch den Knopf K bewegt
werden. Durch diese eigenthümliche Zusammenstellung erhält man bei gekreuzter
Nicol-Stellung ein scharfes, auf eine bestimmte Wellenlänge einstellbares Lichtbild
in Form eines schmalen wagerechten Bandes mit schwarzen senkrechten Mittelstreifen.
Die Bestimmung der Drehung der Polarisationsebene für jede beliebige Farbe
bestimmter Wellenlänge ist sehr genau. Für Licht der D-
bezieh. F-Linie beträgt der Beobachtungsfehler am
Dispersionspolarimeter nur 0,004 bis 0,015°.
Tafeln